理论步骤:
首先通过化学式计算不饱和度 unsaturation=\sum pi - bonds + rings = C + 1 - (\frac{H+X-N}{2}),正常如果不饱和度大于或者等于4,意味着可能存在芳香环。
如果有红外数据,可以看看是否有 C=O, OH, NH, C C, N\equiv C。
从碳谱中确定等价碳原子个数。化学式中有几个碳,谱图中有多少个碳谱峰,确定多少个不等价碳原子。也是判断化学结构式是否有对称性问题。
看1H氢谱谱图,寻找特征指纹谱峰。其中 7.0-8.0ppm 为芳香区域,10.0-11.0 ppm 为醛基区域,12-15ppm 为羧酸区域,而活泼氢一般表现为宽包,羟基和氨基有可能出现在 2-5\ ppm,而酰胺氢在 7.0-8.0ppm 。
同样的也查看 13C 谱图,寻找特征谱峰。
积分氢谱每簇峰,要满足积分面积和分子的氢个数一致。注意,有的活泼氢在质子溶剂中会不出峰,比如羟基在水或者甲醇溶剂中就不出峰。
仔细观察谱峰的裂缝模式,使用N+1规则确定基团片段。
通过氢谱和碳谱确定相应的功能团。
把所有信息整合起来考虑,确定分子结构式链接不会违反 Lewis 规则,考虑上所有的碳原子和氢原子,确定不饱和度是否正确。
实例解析
样品信息
样品编号: 6号
样品分子式: C9H11N
氘代试剂:氘代氯仿(CDCl3)
实验结果
一、碳谱(13C NMR)数据
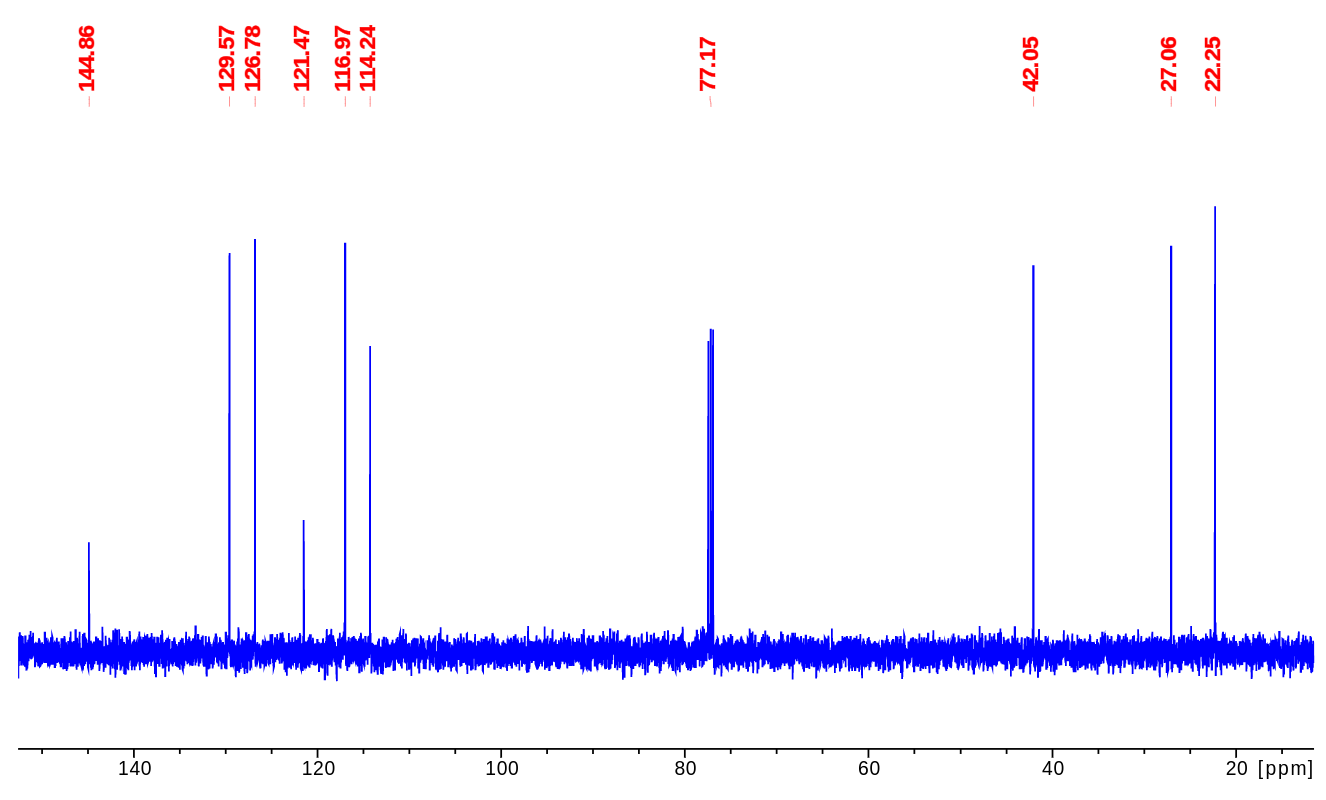
No. | δC /ppm | comment |
1 | 144.86 | Intensity lower |
2 | 129.57 |
|
3 | 126.78 |
|
4 | 121.47 | Intensity lower |
5 | 116.97 |
|
6 | 114.24 |
|
7 | 42.05 |
|
8 | 27.06 |
|
9 | 22.25 |
|
二、氢谱(1H NMR)数据
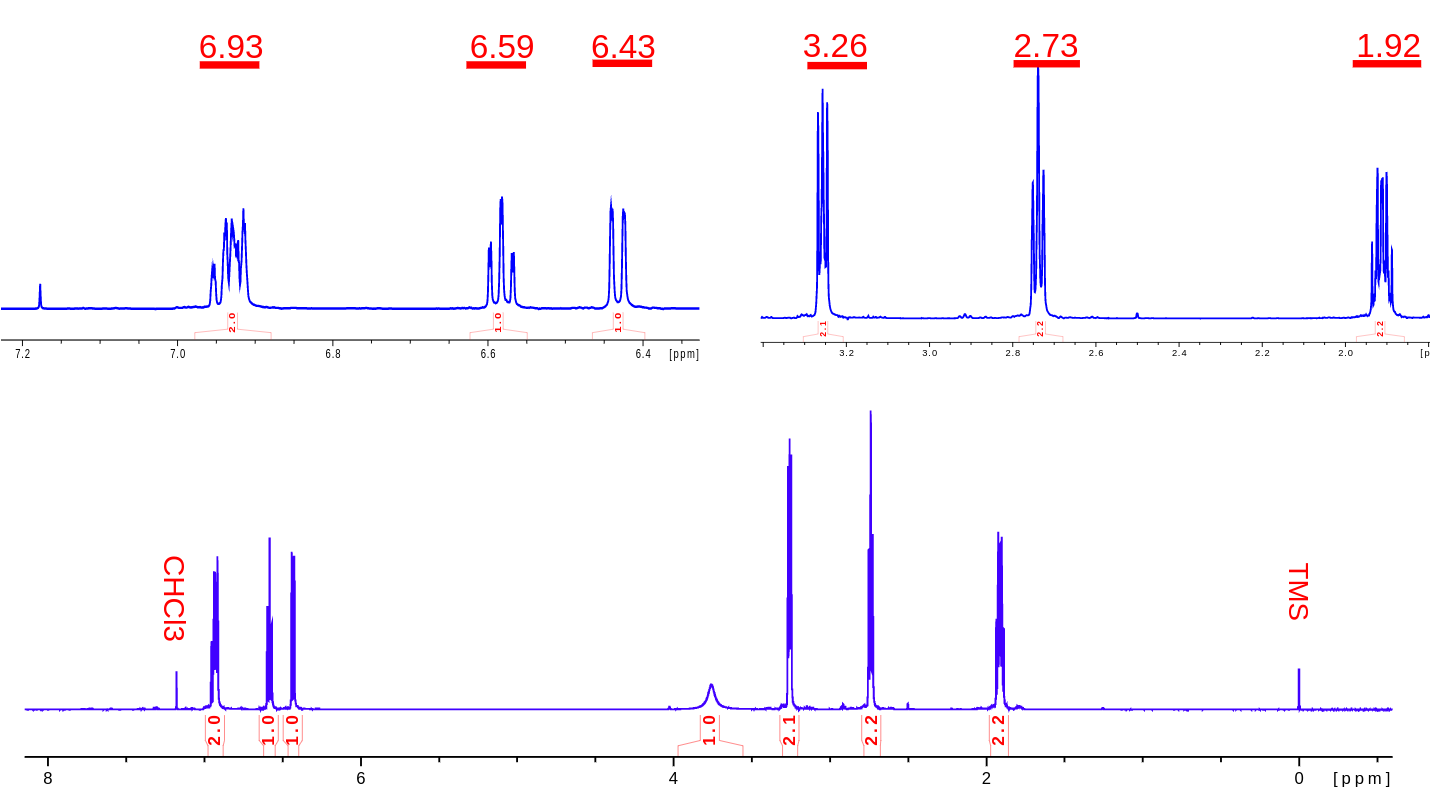
No. | δH /ppm | Integral | J/Hz | Split | Hs |
| comment |
1 | 6.93 | 2.0 |
| m | 2 | CH | overlay |
2 | 6.59 | 1.0 | 7.4 | t | 1 | CH |
|
3 | 6.43 | 1.0 | 7.9 | d | 1 | CH |
|
4 | 3.76 | 1.0 |
| brs | 1 | NH | board peak |
5 | 3.26 | 2.1 | 5.5 | t | 2 | CH2 |
|
6 | 2.73 | 2.2 | 6.5 | t | 2 | CH2 |
|
7 | 1.92 | 2.2 |
| m | 2 | CH2 |
|
结果分析
一、计算不饱和度:(2x9+2-11+1)/2=5
二、13CNMR数据分析
0. 非样品信号:13CNMR约 77ppm的三重峰为溶剂氘代氯仿CDCl3的碳峰。CDCl3的碳因为受到氘(自旋为1)耦合,裂分为三重峰,碳和氘的一键耦合常数为32Hz 【耦合裂分模式】。
1. 目标分子共有9根碳峰,比对分子式C9H11N(有9个碳),说明该分子不具备对称性【化学等价/磁等价】。
2. 碳谱数据显示,芳香区110-150ppm 之间有6个碳峰【双键和芳香区的碳谱特征峰】,比较合理的推断存在一个苯环(占用4个不饱和度)。
三、1H NMR数据分析
0. 非样品信号:内标TMS的化学位移为 0ppm【化学位移定标】,溶剂CDCl3残存氢的化学位移在 7.18 ppm。
1. 选定6.45 ppm(t峰)为积分参考,得到 11 个氢信号,与分子式C9H11N吻合【积分面积】。
2. 芳香区域(6-8ppm)存在4 个氢(信号H1~H3),说明苯环上被取代两个位置。
3. 化学位移 3.76 ppm是一个单重的宽峰(brs),可以推断为活泼氢(H4);结合分子式中有一个N,所以推断存在基团 -NH-【活泼氢核磁性质】。
4. 从积分比看,H5,H6,H7都是亚甲基(CH2);从裂分模式看,三者依次为多重峰(m),三重峰(t),三重峰(t),【耦合裂分模式】所以推断出可能的片段 -CH2-CH2-CH2- (n+1规则,前后两个CH2是t峰,中间CH2是m峰)。
四、数据综合分析
1.目前有一个双取代苯环-C6H4-(没有对称性,所以不会是对位取代),一个氨基 -NH-,片段-CH2-CH2-CH2-:已经有9个碳,11个氢,1个氮,满足分子式。但是不饱和度还差一个,所以考虑存在环。所以合理推断有以下结构:
(a) (b)
(b)
2.比对双取代苯环的氢耦合裂分情况:
结构(a):氢谱芳香区应该是两个d峰,两个t峰(苯环邻位氢的耦合常数差不多)【思考两个双重峰和三重峰的关系】。
结构(b):氢谱芳香区应该是两个d峰,一个t峰,一个s峰。
实际谱图中,氢谱芳香区(高场到低场)依次为d峰,t峰,m峰(2组H)重叠。所以结合谱图倾向于结构(a),低场m峰为一个d峰和一个t峰的重叠。
五、结构(a)的碳/氢谱峰归属
1.13C NMR数据:
低场芳香区110-150ppm 之间是苯环6个碳的碳峰,高场其余三个碳峰对应片段 -CH2-CH2-CH2-。
由于9号位连接着电负性较强的氨基,所以会偏向低场,9号位碳峰应该在42.05ppm【诱导效应】;7号位连着苯环,相对的碳谱也会往低场偏,所以碳峰27.06ppm对应7号位碳【超共轭效应】;于是最高场的22.25ppm就是8号位碳谱信号。
考察苯环芳香区的碳信号归属。1号位季碳相应的化学位移会往低场偏,而且1号位直接与N相连接,所以1号位碳更低场为144.86ppm【诱导效应】。6号位虽然也是季碳,如果没有NH基团和苯环形成的p-π共轭给电子效应的影响,该位置的季碳化学位移将会向低场偏移10ppm左右;但是受到共轭效应,导致其化学位移向高场移动,考虑到季碳的谱峰信号强度相对较弱,所以推断6号位碳峰为121.47ppm。接下关于2,3,4,5号位碳的归属:考虑NH基团和苯环形成的p-π共轭效应是给电子效应,会引起苯环化学位移往高场偏移,偏移最大应是邻位2号位,其次对位4号位。所以指认2号位碳峰为114.24ppm,4号位为116.97ppm。那么3和5号位的核磁信号谁更高场呢?7号位的亚甲基会对苯环形成超共轭效应,而且是给电子效应,所以5号位的化学位移应该会偏向高场一点。
2. 1H NMR数据:
三个亚甲基片段-CH2-CH2-CH2-:同碳谱分析一样,连接NH基团的氢化学位移也会往低场偏移,所以3.26ppm 定为9号位;连接苯环的位置也会低场偏移,所以2.73ppm定为7号位氢,剩下1.92ppm为8号位氢。裂分模式:9号为t峰(其中NH由于活泼氢导致弛豫快,对邻位并没有形成有效的耦合,但是该位置并不是完美的三重峰),7号氢谱也是t峰,8号为m峰。
苯环氢分析:和碳谱分析同理,NH基团邻位的2号位氢处于最高场6.43ppm, NH基团对位的4号位氢为6.59ppm,3,5号位氢比较临近重叠在6.93ppm。从裂分模式上也可辅助证明以上推断。
综上讨论,可得:
No. | δC /ppm | δH /ppm |
1 | 144.86 | ---------- |
2 | 114.24 | 6.43(d,1H) |
3 | 129.57 | 6.93(m,1H) |
4 | 116.97 | 6.59(t,1H) |
5 | 126.78 | 6.93(m,1H) |
6 | 121.47 | ---------- |
7 | 27.06 | 2.73(t,2H) |
8 | 22.25 | 1.92(m,2H) |
9 | 42.05 | 3.26(t,2H) |
六、化学位移预测
1.将(a)化学结构式输入到nmrdb.org网站进行预测
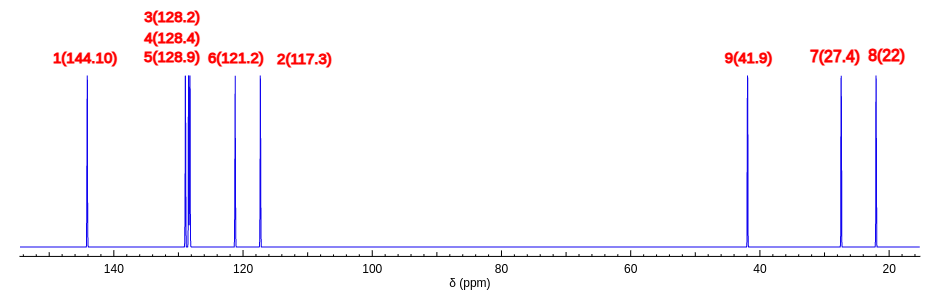
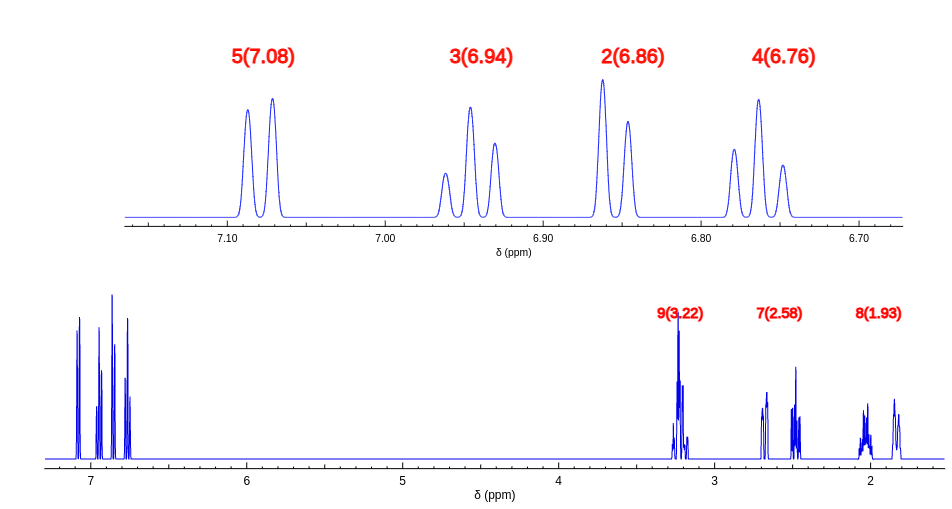
从预测的碳谱信息可以看出来,除了4号位置的化学位移和我们推导的不一样。其他位置碳谱信息还是比较吻合的。4号位的碳谱的特殊性来源于NH对苯环形成的共轭效应的特殊性。
而氢谱的预测与实际测试谱图,在亚甲基这个区域化学位移位置没有问题,但是裂分模式差别很大。可能时因为预测考虑到NH的耦合裂分,还有亚甲基上面两个氢的强耦合情况。但是实际测试中NH并没有表现出来对相邻的亚甲基的耦合,并且三个亚甲基上面的两个氢也没有表现出强的耦合特性。对于苯环区域的5,3,实际测试它们差别没有那么大,并且测试数据中2氢谱化学位移比4低场,和预测不一致。
2.将(b)化学结构式输入到nmrdb.org网站进行预测


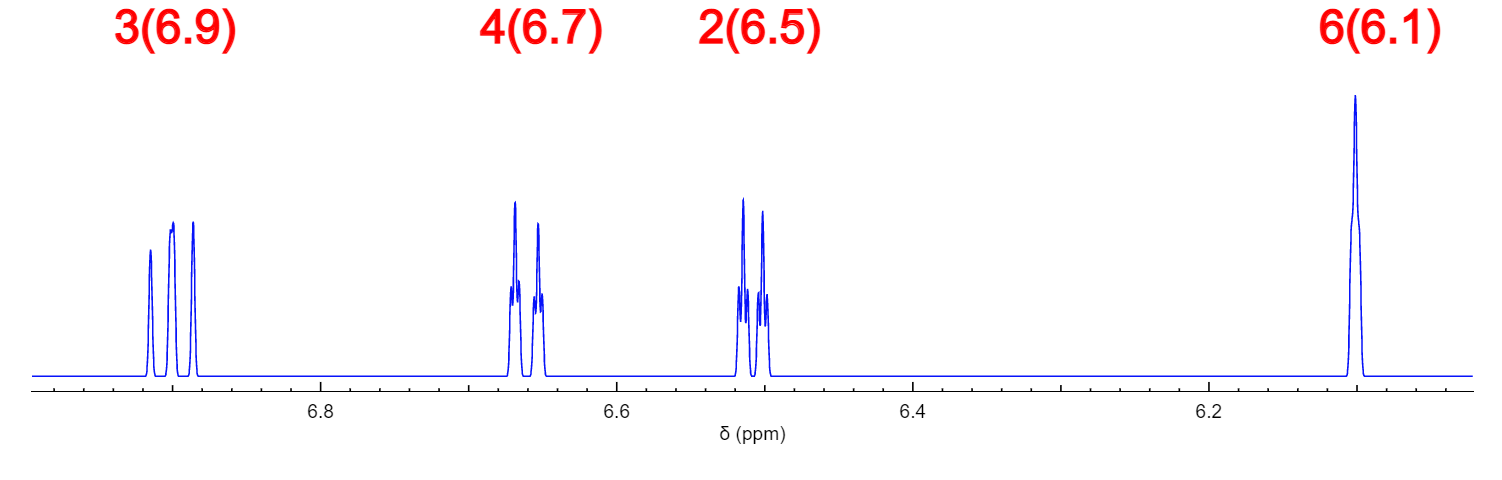
在氢谱的苯环区域有一个很明显的单重复在高场。并且苯环区域的裂分模式有一个三重峰在最低场,两个双重峰和一个单重峰。而碳谱角度来说,1和5都是季碳,并且它们在最低场位置,也和实验谱图不吻合。
